Structure-Based Drug Discovery

Target-Based approaches complement the drug-discovery methods at the ITR
The scientific advances in biochemistry and structural biology during the second half of the 20th century allow us to approach drug discovery research at the level of the proteins/enzymes that regulate the biological process in the organisms.
Structure Based Drug Discovery at ITR
The rotating image shows the tetramer of the FBPases protein from Francisella tularensis, an important pathogen. FBPase (Fructose-1,6-Bisphosphatase) is a key enzyme in the gluconeogenic pathway. This pathway synthesizes Glucose from other carbon sources, e.g. glycerol, and is critical for the survival of the pathogen in the host.
Target Based
Fig1a.
Advances in biochemistry and genetics in the first half of the 20th century established that most biological processes were controlled by metabolites synthesized in metabolic cascades or pathways. Thus, the active compound (M) is synthesized via a metabolic cascade (A>B>C…. M). The synthesis of those compounds is controlled (driven) by enzymes (E1, E2, E3…). Inhibition of any of those enzymes would prevent the synthesis of the active compound M.
Structure based
Fig1b.
The protein of interest is crystallized possibly in the presence of a ligand (A, e. g. inhibitor) bound and the structure determined by X-ray crystallography. Analysis of the bound ligand A (i.e. molecular modeling) permits the study the inter-molecular interactions between the target and the ligand, from which a hypothetical superior compound B is designed, synthesized and characterized. Ligand B is also crystallized bound to the enzyme and the process is repeated. After several cycles, an optimized compound can be considered for further development, possibly leading to a drug candidate.
more continued information
Currently, this methodology is being used at the ITR to identify, validate and optimize compounds for two targets of interest for anti-TB therapy:
ClpC1: Important target for protein homeostasis. This target is essential for the survival of the M. tuberculosis pathogen. The structures of two potent natural macro-cyclic peptides Rufomycin and Ecumicin bound to the N-terminal domain of ClpC1 (i.e. ClpC1-NTD) have been characterized. Fig. 2a,b.
FBPasesII: Key enzyme in the gluconeogenesis pathway of many organisms. This is an exploratory target. Fig.3. View of the active site with the key elements for catalysis color coded: PO4-6 binding, Arg165-Pro166-Arg167 (cyan); substrate F1,6BP binding, Asp187-Gly188-Asp189 (green); Helical N-term, PO4-1 binding site Thr89xxxLys94 (blue); and metal Mn2+ binding site, Asp84—Glu87 (dark grey). Substrate Fructose-1,6-bisphosphate (green).
Figure 2a
Figure 2a
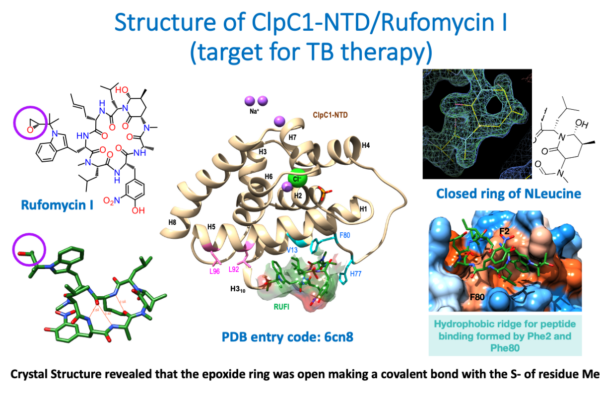
Figure 2a
Description of Figure 2a
Fig.2a.
The images summarize the crystallographic results derived from the structure of the cyclic peptide Rufomycin (left, top and bottom), complexed with the N-terminal domain (NTD) of the target ClpC1 (center). The structure was determined at a resolution of 1.4 Å (high resolution) and the electron density map (right, top) showed unambiguously the conformation of the closed N-leucine ring. (Right, bottom) show the binding pocket of the Rufomycin cyclo-peptide on the surface of the ClpC1-NTD. Surprisingly, the epoxide ring shown in the 2D sketch of Rufomycin (purple circle, top left) was found open in the crystal structure, making a covalent bond with the SD atom of the N-terminal Methionine 1. The structure and the crystallographic data have been deposited in the PDB with accession code 6cn8.
For further details the complete publication on the structure of ClpC1-NTD-Rufomycin complex can be found in the attached file [PDF].
Figure 2b
Figure 2b
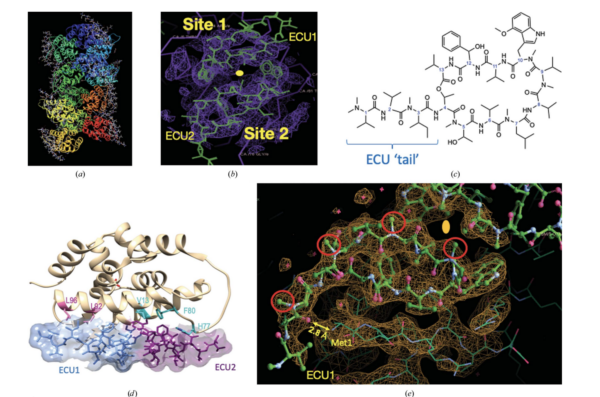
Figure 2b
Figure 2b
Fig. 2b.
The most striking insight of this structure is that the Ecumicin tri-decapeptide (13 a.a.) binds to the target in a 1:2 stoichiometry (1 target: 2 Ecumicin).
a) Crystal packing of the 12 molecules of ClpC1-NTD and the corresponding 24 Ecumicin molecules in the crystal structure.
b) Electron density of the two sites related by a 2-fold axis domain repeat in the ClpC1-NTD.
c) ChemDraw 2D rendering of the Ecumicin tri-decapeptide showing the extended tail.
d) The two molecules of Ecumicin (ECU1-ECU2) related by the 2-fold dyad running vertical in the plane of the paper.
e) Detail of the electronic density of the crystal structure showing the interaction between the N-Terminal Methionine1 and a carbonyl (C=O) group of the Ecumicin tail in the ECU1 site.
Further details of the structure can be found in the below PDF.
Figure 3
Figure 3
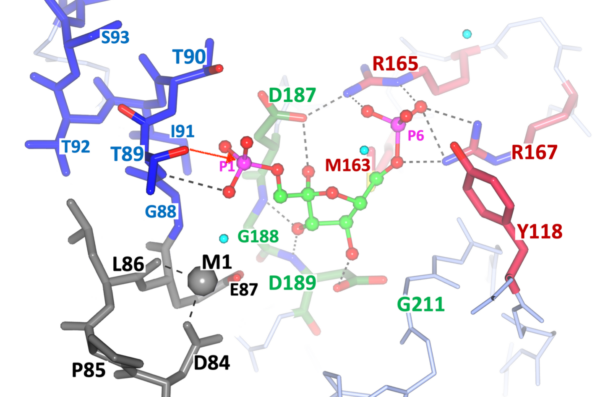
Figure 3
Figure 3
Fig. 3.
This Figure correspond to the most recent crystallographic results of the other target under study at ITR, namely Fructose-1,6-Bisphosphatase (FBPase) of important pathogens.
The project is focused on the FBPases Class II of two important pathogens: M. tuberculosis and F. tularensis. The structures of this target from M. tuberculosis and F. tularensis have been solved and refined at ITR. Our most recent publication suggests and novel catalytic mechanism for the entire class and the structural details are highlighted in Fig. 3. The structural elements for the catalytic cleavage of Phosphate 1 (P1, left side) are color coded: ii) green center, substrate F1,6BP binding with the conserved motif Arg165-Pro166-Arg167 anchoring the uncleaved phosphate; ii) Grey Metal 1 binding pocket (Mg++, or Mn++); and iii) Thr89-OH nucleophile and the helical N-terminal stabilizing the transition state. The red arrow from the Thr89-OH suggest the nucleophilic attack to the leaving phosphate (P1).
Further details can be found in the two most recent publications on the structures of FtFBPasesII from F. tularensis with two different metal cofactors (Mg2+, Mn2+).
The previous papers on the target validation and the structure of M. tuberculosis FBPase class II (MtFBPaseII)
Can be found below: